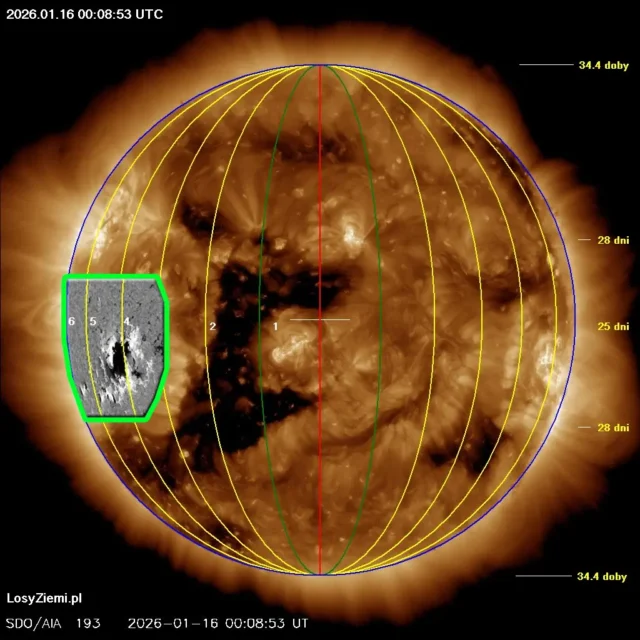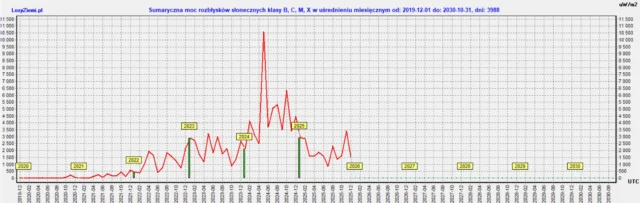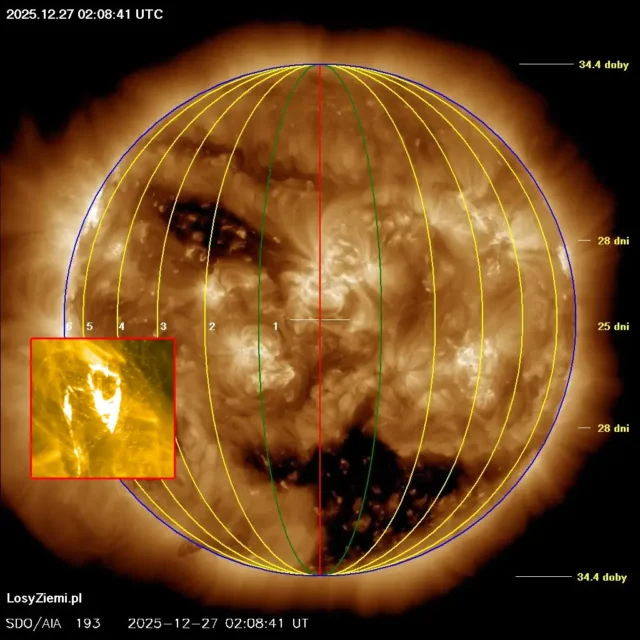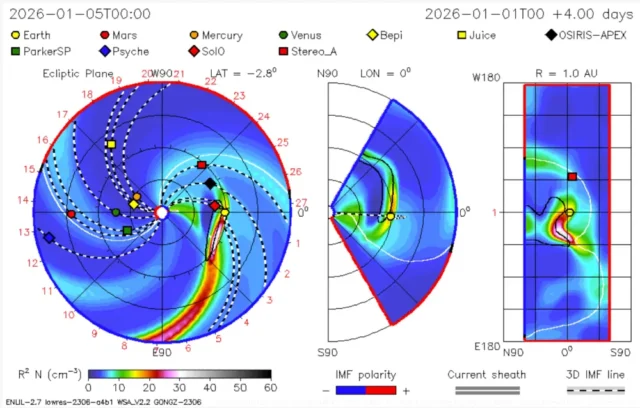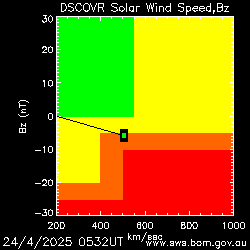
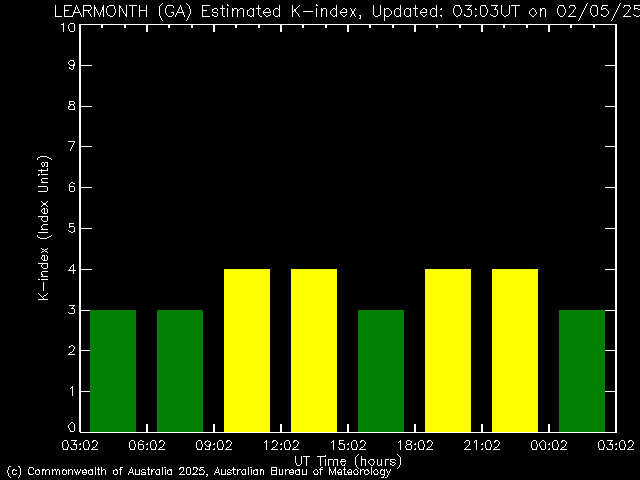
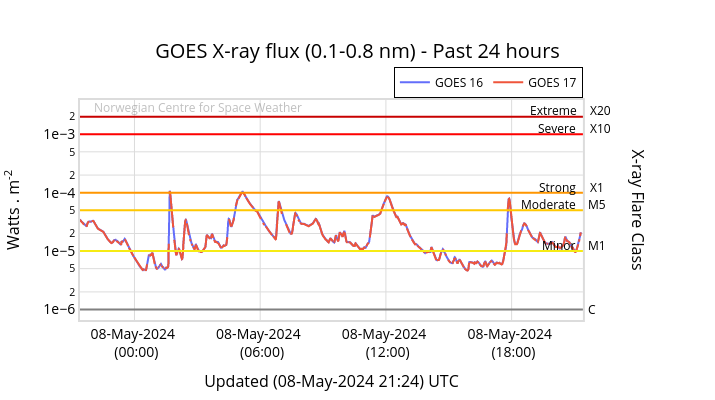
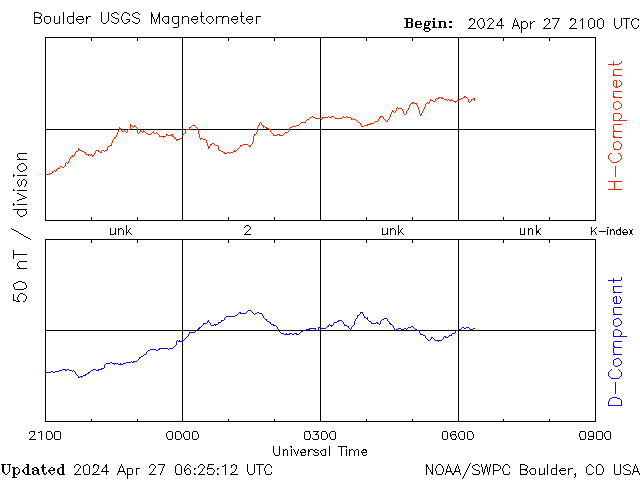


◄ Treść dostępna po zalogowaniu. ►
◄ Zaloguj się. ►
Trwa zbiórka darowizn na utrzymanie serwisu, więcej informacji po prawej stronie.
Zasady wsparcia finansowego serwisu, jak również możliwości otrzymania konta opisane są TU.


W czerwcu zebraliśmy:
Szybka wpłata anonim.
z czego cała reszta czytelników spoza Grupy VIP: 1.4%